The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3
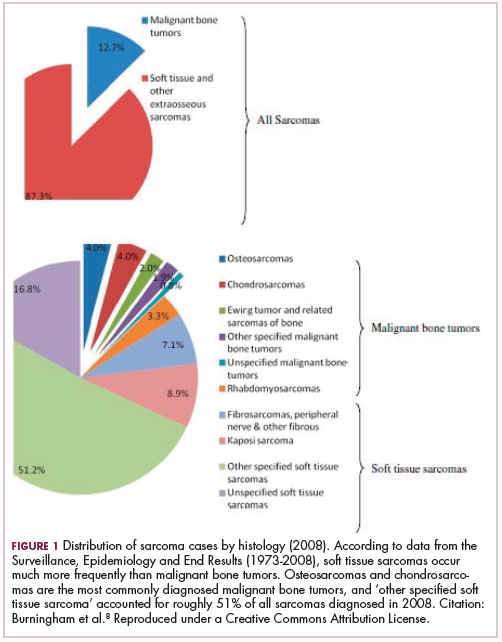
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).

Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
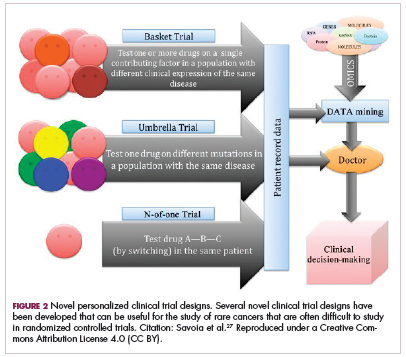
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44